24小时服务热线:19103801095
NEWS CENTER
Recommend case
contact us
本文介绍了美国医疗器械市场准入类型、分类方法、法规介绍及注册流程。建议收藏学习。

FDA是美国最资深的综合性消费者保护的政府机构,主要是促进以及保护健康,监管领域涵盖食品,药品,生物制品,化妆品,动物和兽医药物,还有烟草,旗下的CDRH部门监管医疗器械和放射性产品,评估医疗器械上市前后的安全与有效性以及为病患和供应商提供及时连续的通道。
1、医疗器械市场准入类型
510k
510k为低、中风险器械的市场应用新器械(New Device)与合法销售器械(Predicate Device)之间的“实质等同(SE)”,通过预期用途,器械的功能,性能测试进行对比。
实质等同性比较是要证明所申请上市的产品和已在美国市场上合法销售的产品在安全性和有效性方面比较是实质相等的。选择合适的产品进行比较是510(K)申请的关键步骤。在进行比较时应从如下方面进行考虑:
企业必须提供充足的资料证明,所申请上市的器械和被比较的器械是实质相等的(SE),否则510(K)申请不奇亿娱乐过。

PMA
PMA是FDA监管最严格的III类医疗器械的上市前提交类型,申请PMA,也就是III类医疗器械的注册申请,必须充分证明其安全性和有效性,通过IDE申请临床试验,提交大量的研发文档和技术文档。
HDE
人道主义器械豁免可用于疾病或者少于每年8000人的情况—如罕见的右心衰竭。他们可以在没有临床证据的情况下得到豁免,申请HUD再由HDE审批,但是申请人必须对产品安全性作出解释并使人相信其应用利大于弊。
De Novo
De Novo(产品风险等级的重新分类)是一种基于风险的分类过程。对于没有合法上市对比产品的新型医疗器械,即使是中低风险,仍无法通过510(k)申请建立实质等同(SE)从而获得上市许可。针对这类产品,FDA建立了De Novo申请途径。
De Novo使得更多符合奇亿娱乐性能的新型器械上市,并可以作为510 (k)申请路径实质等同评价的对比产品。同时,FDA也会采取新方法,促进在510(k)申请路径的实质等同对比过程中使用更加奇亿娱乐的对比产品,从而又促进更多的医疗器械采用De Novo途径。
IDE
IDE指的是试验用器械豁免,是FD&CAct对于仅用于临床试验的医疗器械的管制措施,其主要的精神是让研究发展中的医疗器械可以免除掉对于以上市销售为目的的器械产品的种种管制,而以较简单的方式让制造奇亿娱乐过临床试验来收集安全性和有效性的信息资料,从而为510(k)和PMA申请提供数据支撑。FD&CAct 520(g)条款中授权FDA对临床试验用的器械,可以免除企业注册、产品登记、标示、510(k)、PMA或医疗器械伤害报告等规定,除设计控制外,也不必遵循质量体系法规的要求,但仍要具有与上市时同样的合格标准(也就是说,制造商必须确保临床试验用的医疗器械已经符合安全性评估),遵守特别的标识规定(如注明限临床试验使用或警告语),制造商也不准对临床试验用的医疗器械有广告、商业化或任意延长临床试验期限的行为。对于大多数的PMA申请而言,临床试验要求是必须的。然而,制造商在提交510(k)申请的时候,却仅在少数情况下要求提供临床试验数据。这里“试验”还包括对已合法上市器械的改进和新用途的临床评价。除豁免的情况外,所有试验用器械在其临床试验启动前都必须取得IDE许可。

2、医疗器械分类及其查询方法
分类依据
FDA医疗器械的分类主要取决于它的预期用途和适应症。例如,手术刀的用途是切组织。当器械的标签上增加了更专业的适应症,如“用于切割角膜”,就会出现预期用途的子集,即适应症。
(1)Based on device description and intended use
(2)Determines extent of regulatory control
(3)Class I, II, or III
– increases with degree of risk
(4)Product Codes: three-letter coding to group similar devices and intended use
分类查询
要查找器械的分类,以及是否存在任何豁免,需要查找 regulation number。
有两种方法来实现:
1. 直接进入分类数据库,搜索一个器械名称的一部分,或者,如果你知道器械的组别device panel,直接转到该panel,鉴别你的器械和相应的监管类型。
2. 如果你已经知道相应的panel,也可以直接访问CFR,并通过阅读分类器械的列表来找到该器械的分类,或者如果不确定,可以使用产品代码分类数据库中的关键字目录。在大多数情况下,该数据库将识别CFR中的分类规则。
如何查询?
例如:额温枪
步骤一:输入产品名称。

步骤二:点击Search后,然后出现下面的页面,这个页面其实已经可以看出器械分类了,我们再点Thermometer,Electronic,Clinical,看里面更详细的信息。

步骤三:除了可以看出分类,还可以找到该产品的product code,法规,上市提交类型,是否GMP豁免,是否可以第三方审查(Third party review)等。

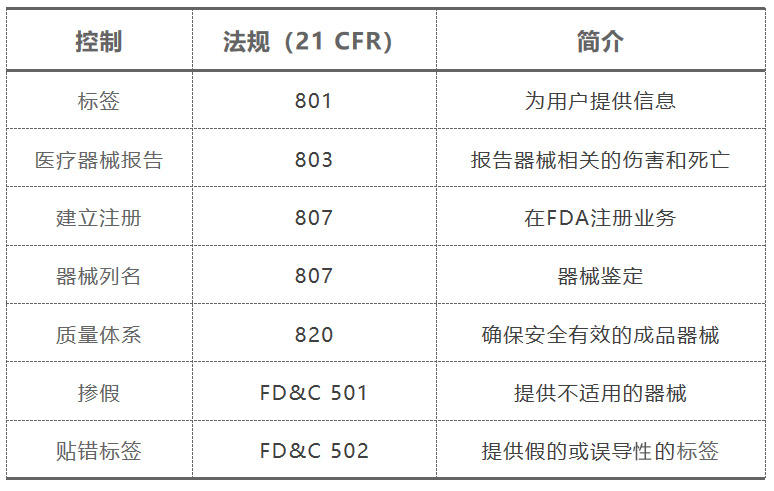
3、法规简介
CFR中有关医疗器械的法规:
(1)21 Code of Federal Regulations (CFR): Parts 800-1050
– 800-861: cross-cutting device requirements
(2)Example: 812 - Investigational Device Exemption
– 862-1050: device-specific requirements
(3)Example: 876 - Gastroenterology and Urology Devices(肠胃科和泌尿科器械)
(4)21 CFR: Parts 1-99
– general medical requirements that also apply to medical devices
常见的法规举例:
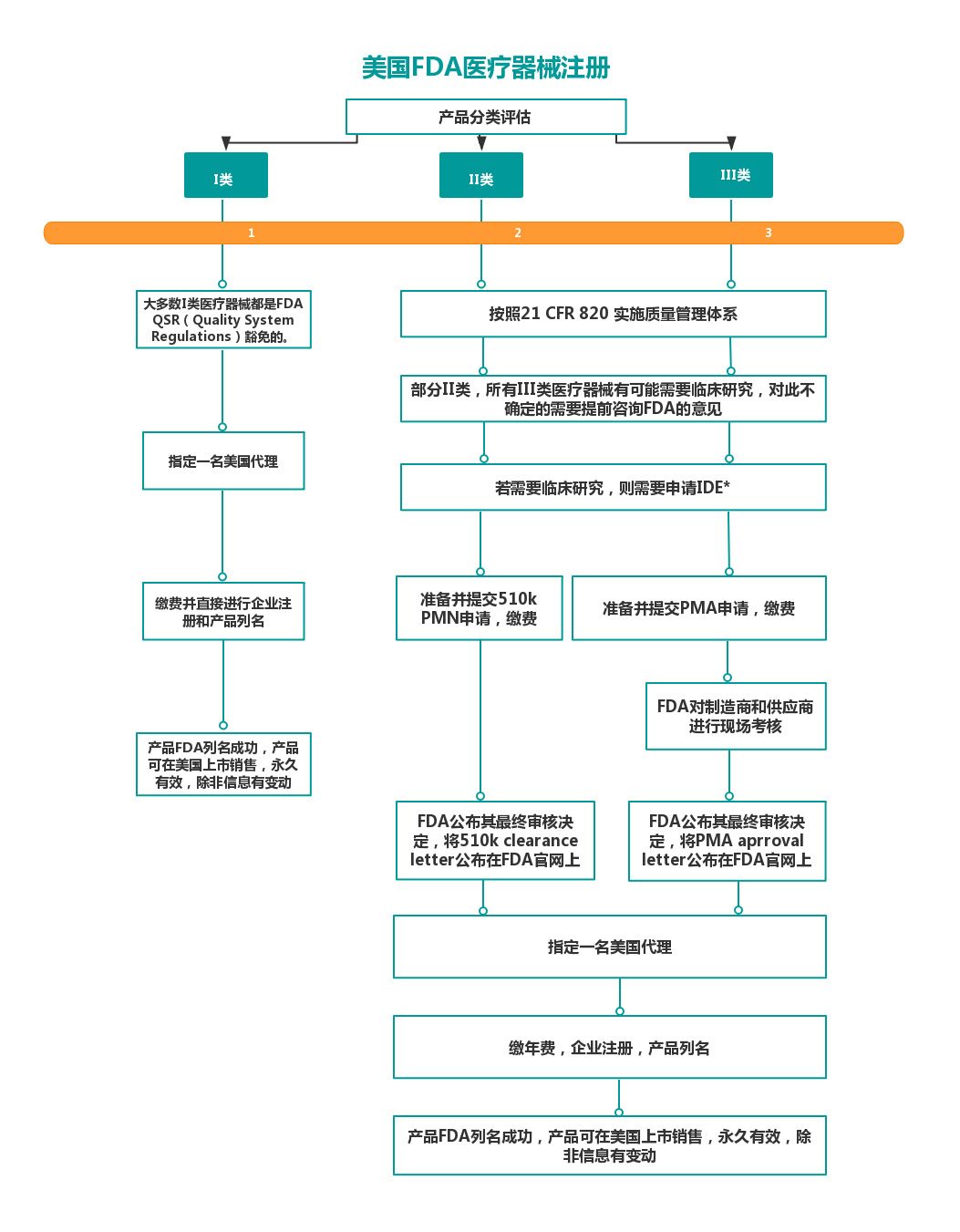
4、医疗器械注册流程
站点声明:
本网站所提供的信息仅供参考之用,并不代表本网赞同其观点,也不代表本网对其真实性负责。图片版权归原作者所有,如有侵权请联系我们,我们立刻删除。如有关于作品内容、版权或其它问题请于作品发表后的30日内与本站联系,本网将迅速给您回应并做相关处理。
奇亿娱乐(中国)有限公司专注于医疗器械、诊断试剂产品政策与法规规事务服务,提供产品注册申报代理、临床合同(CRO)研究、产品研发、GMP质量辅导等方面的技术外包服务。










